Die Pathologie im Rahmen des europäischen und nationalen Medizinprodukterechts: Betrieb, Anwendung und Eigenherstellung von In-vitro-Diagnostika
Andy Kahles 1Hannah Goldschmid 1
Anna-Lena Volckmar 1
Daniel Kazdal 1
Ulrich M. Gassner 2
Michael Vogeser 3
Monika Brüggemann 4
Karl-Friedrich Bürrig 5
Vanessa Kääb-Sanyal 5
Christa Flechtenmacher 1
Peter Schirmacher 1
Albrecht Stenzinger 1
1 Pathologisches Institut, Universitätsklinikum Heidelberg, Deutschland
2 Juristische Fakultät, Universität Augsburg, Deutschland
3 Institut für Laboratoriumsmedizin, LMU Klinikum, LMU München, Deutschland
4 Klinik für Innere Medizin II, Sektion für Hämatologische Spezialdiagnostik, Universitätsklinikum Schleswig-Holstein, Kiel, Deutschland
5 Berufsverband Deutscher Pathologinnen und Pathologen e.V., Berlin, Deutschland
Zusammenfassung
Institute und Praxen für Pathologie fungieren als Betreiber, Anwender und Eigenhersteller von In-vitro-Diagnostika und unterliegen funktionsabhängig nationalen und europäischen Regularien. Durch das Inkrafttreten der EU-Verordnung über Medizinprodukte (Verordnung (EU) 2017/745, MDR) und der EU-Verordnung über In-vitro-Diagnostika (Verordnung (EU) 2017/746, IVDR) ergab sich ein regulatorischer Anpassungsbedarf im deutschen Medizinprodukterecht. Damit wurde ein neuer Rechtsrahmen geschaffen, in dem sich Institute und Praxen für Pathologie je nach ihrer Funktion als Anwender, Betreiber oder Eigenhersteller von In-vitro-Diagnostika bewegen. Diese Zusammenstellung der aktuellen Rechtslage stellt eine Momentaufnahme dar und bietet einen aktuellen Überblick über die Landschaft des Medizinprodukterechts.
Schlüsselwörter
IVDR, Qualitätsmanagement, Gesetzgebung, regulatorische Anforderungen, Gesetze und Verordnungen, In-vitro-Diagnostika (IVD)
Einleitung – die Pathologie im Rahmen des Medizinprodukterechts
Durch die Einführung der In-vitro-Diagnostika-Verordnung (Verordnung (EU) 2017/746, IVDR) und der Medizinprodukteverordnung (Verordnung (EU) 2017/745, MDR) ergab sich im deutschen Medizinprodukterecht der Bedarf einer regulatorischen Anpassung. Somit wurde ein neuer europäischer und nationaler Rechtsrahmen geschaffen, in dem sich Institute für Pathologie bewegen. Mit diesem Artikel möchten wir einen Überblick über die Landschaft des Medizinprodukterechts geben, die Pathologie darin verorten und eine Orientierungshilfe für die Praxis geben.
In der Pathologie führen ärztlich verantwortete Prozessketten in der Analytik zu einem validen Befund, der eine Diagnose und eine bestmögliche Krankenversorgung ermöglicht (Abbildung 1a [Abb. 1]). Bei jedem Glied in der Prozesskette können dabei verschiedenartige In-vitro-Diagnostika (IVD) einzeln oder in Kombination miteinander zum Einsatz kommen. In-vitro-Diagnostika sind gemäß der definierten Begriffsbestimmungen innerhalb der beiden Verordnungen (EU) 2017/746 (IVDR) und (EU) 2017/745 (MDR) eine Teilmenge der Medizinprodukte und unterliegen folglich dem europäischen und nationalen Medizinprodukterecht (Abbildung 1b + c [Abb. 1], Begriffsbestimmungen siehe Tabelle 1 [Tab. 1]) [17], [18]. Hiervon ausgenommen und abzugrenzen sind Produkte des allgemeinen Laborgebrauchs, für die der Hersteller keine spezifische medizinische Zweckbestimmung erklärt [26]. Diese sind aber ebenfalls essenzieller Bestandteil in der Prozesskette zur optimalen Diagnosefindung.
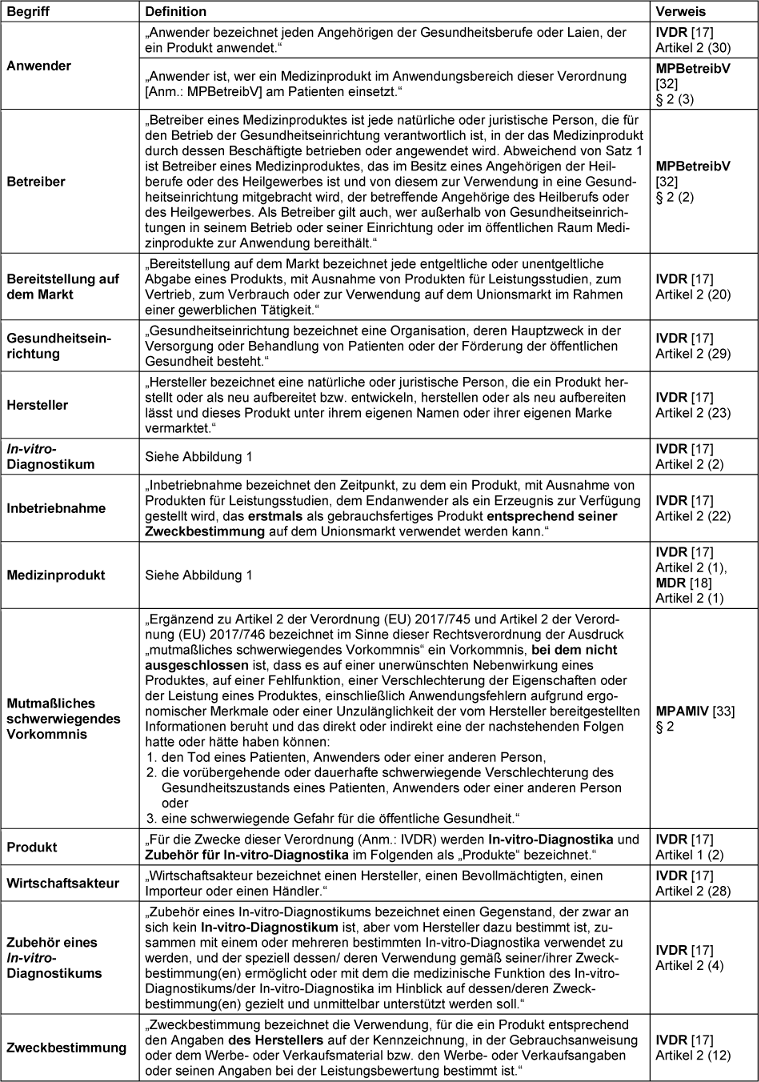

Institute und Praxen für Pathologie agieren aus juristischer Sicht als Betreiber und Anwender von In-vitro-Diagnostika (Begriffsbestimmung ‚Betreiber‘ und ‚Anwender‘ siehe Tabelle 1 [Tab. 1]). Bringt der Hersteller auf einem IVD eine CE-Kennzeichnung an, erklärt er damit die Konformität mit geltendem europäischem Recht. Je nach Risikoklasse seines IVD-Produktes darf der Hersteller das CE-Kennzeichen erst nach erfolgreicher Prüfung durch eine privatwirtschaftlich agierende Konformitätsbewertungsstelle (sogenannte „Benannte Stelle“) anbringen. Die CE-Kennzeichnung ist Voraussetzung für das Inverkehrbringen auf dem europäischen Markt. Setzen Institute und Praxen für Pathologie IVD zur Diagnosefindung ein, die diese CE-Kennzeichnung nicht aufweisen (z.B. Research-use-only(RUO)-Produkte oder selbst entwickelte Produkte) oder setzen sie IVDR-konforme IVD (CE-IVD) außerhalb ihrer vom Hersteller gegebenen Zweckbestimmung oder abweichend ihrer Gebrauchsanweisung ein, werden sie selbst zum (Eigen-)Hersteller sogenannter In-house-in-vitro-Diagnostika (IH-IVD) und übernehmen einige Herstellerpflichten gemäß Artikel 5 (5) der IVDR [25], [26], [30], [31] (Begriffsbestimmung ‚Hersteller‘ siehe Tabelle 1 [Tab. 1]). Dieses Vorgehen ist vom Verordnungsgeber ausdrücklich erlaubt und erwünscht. In Erwägungsgrund 29 der Präambel der IVDR wird die besondere Bedeutung von Gesundheitseinrichtungen und den von ihnen selbst entwickelten IVD hervorgehoben. Mit der Konkretisierung des Erwägungsgrunds durch Artikel 5 (5) der IVDR erkennt der Unionsgesetzgeber den Nutzen und die Notwendigkeit von IH-IVDs an und ermöglicht deren Einsatz für eine optimale Patientenversorgung [26] (siehe auch Steckbrief IVDR in Tabelle 2 [Tab. 2]).

Institute und Praxen für Pathologie können folglich multifunktional als Betreiber und/oder Anwender, aber auch als Eigenhersteller von IVD tätig sein. Das europäische und nationale Medizinprodukterecht gibt hierfür einen Rechtsrahmen vor, innerhalb dessen sich Institute für Pathologie und deren Beschäftigte in Abhängigkeit ihrer jeweiligen Funktion bewegen.
Anpassungen im Medizinprodukterecht durch die Einführung der MDR und IVDR
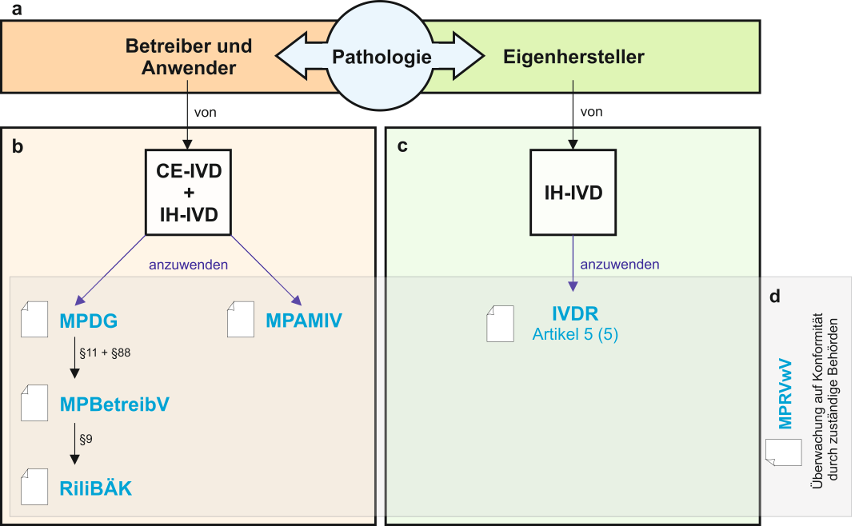
Durch die Einführung der beiden EU-Verordnungen IVDR und MDR sollen unionsweit harmonisierte Rahmenbedingungen geschaffen werden, um ein hohes Maß an Gesundheitsschutz für Patienten und Anwender zu gewährleisten. Gleichzeitig soll ein reibungslos funktionierender Binnenmarkt für Medizinprodukte und In-vitro-Diagnostika sichergestellt werden (Steckbriefe IVDR und MDR siehe Tabelle 2 [Tab. 2] und Tabelle 3 [Tab. 3]) [25]. EU-Verordnungen sind verbindliche Rechtsakte, die mit ihrem Geltungsbeginn unmittelbar und einheitlich in allen Unionsstaaten gelten, ohne dass sie in nationales Recht umgesetzt werden müssen. Dennoch sind im nationalen Medizinprodukterecht umfängliche Anpassungen erforderlich und auch erlaubt, um beispielsweise Zuständigkeiten, Überwachungsmaßnahmen, Strafen und nationale Anforderungen, die allerdings nicht im Widerspruch zu den EU-Verordnungen stehen dürfen, festzulegen (Abbildung 2 [Abb. 2]).


Die Anpassung des nationalen Medizinprodukterechts an die neuen EU-Verordnungen wird durch das Medizinprodukte-EU-Anpassungsgesetz (MPEUAnpG) umgesetzt [21]. Darin enthalten ist das Medizinprodukterecht-Durchführungsgesetz (MPDG, Artikel 1 im MPEUAnpG), welches das bisherige Medizinproduktegesetz (MPG) ersetzt (Abbildung 2 [Abb. 2]). Im Unterschied zum abgelösten MPG handelt es sich beim neuen MPDG um ein Begleitgesetz, das die beiden EU-Verordnungen MDR und IVDR um nationale Vorgaben ergänzt.
MPDG – das Medizinprodukterecht-Durchführungsgesetz
Mit dem „Gesetz zur Durchführung unionsrechtlicher Vorschriften betreffend Medizinprodukte (Medizinprodukterecht-Durchführungsgesetz, MPDG)“ [22] wird das nationale Medizinprodukterecht an die neuen unionsrechtlichen Vorschriften angepasst und das bisherige Medizinproduktegesetz (MPG) abgelöst (Abbildung 2 [Abb. 2], Steckbrief MPDG siehe Tabelle 4 [Tab. 4]). Das MPDG gilt auch für Produkte im Anwendungsbereich der IVDR und gilt somit auch für Institute und Praxen für Pathologie, die als Eigenhersteller, Betreiber und Anwender von IVD agieren können (Abbildung 3 [Abb. 3]). Das MPDG regelt die Straf- und Bußgeldvorschriften bei Verstößen gegen die IVDR und gegen das MPDG. Institute und Praxen für Pathologie unterliegen der allgemeinen Marktüberwachung durch die zuständigen Aufsichtsbehörden (Kapitel 5, § 77). Sie dürfen nur mängelfreie IVD betreiben und anwenden, um Patienten, Anwender und mögliche Dritte nicht zu gefährden (§ 11, Satz 1). Darüber hinaus müssen sie die Medizinprodukte-Betreiberverordnung (MPBetreibV) einhalten (§ 11, Satz 2). Die Pflichten für Institute für Pathologie als Betreiber und Anwender von IVDs sind daher primär in der MPBetreibV festgelegt.

MPBetreibV – die Medizinprodukte-Betreiberverordnung
Die „Verordnung über das Errichten, Betreiben und Anwenden von Medizinprodukten (Medizinprodukte-Betreiberverordnung, MPBetreibV)“ war bereits seit 1998 eine nationale Rechtsverordnung zum Medizinproduktegesetz (MPG). Da das MPG auf Grundlage des MPEUAnpG durch das MPDG ersetzt wurde, wurde auch die MPBetreibV überarbeitet [32], zuletzt im April 2021. Aktuell sieht ein Referentenentwurf des Bundesgesundheitsministeriums eine mögliche geplante Änderung der MPBetreibV vor [10].
Die MPBetreibV setzt § 11 des MPDG in Verbindung mit § 88 Abs. 1 Satz 1 Nr. 6 a), b) und c) um. Sie gilt für das Betreiben und Anwenden von Medizinprodukten und In-vitro-Diagnostika, einschließlich der damit zusammenhängenden Tätigkeiten, wie z.B. der Einrichtung, der Instandhaltungsmaßnahmen und regelmäßigen Prüfungen und beschreibt die dafür geltenden Betreiber- und Anwenderpflichten (Steckbrief MPBetreibV siehe Tabelle 5 [Tab. 5]).

Die MPBetreibV legt fest, wie Einrichtungen, welche laboratoriumsmedizinische Untersuchungen durchführen, als Betreiber von IVDs zu verfahren haben, um eine sichere und ordnungsgemäße Anwendung zu gewährleisten. Dazu legt die MPBetreibV sowohl Betreiber- als auch Anwenderpflichten fest, die für Institute und Praxen für Pathologie und deren Personal relevant sind. Ziel ist es, eine sichere und ordnungsgemäße Anwendung von Medizinprodukten innerhalb der Gesundheitseinrichtung sicherzustellen. Medizinprodukte, und damit auch IVDs, dürfen nur entsprechend der Zweckbestimmung und nur durch kompetentes und entsprechend eingewiesenes Personal angewendet werden (§ 4, § 5). Eine Ausnahme hiervon – die mögliche Anwendung abweichend von der Zweckbestimmung des Herstellers – zeigt die IVDR auf. Gesundheitseinrichtungen dürfen von der Zweckbestimmung des Herstellers abweichen, um auf die spezifischen Bedürfnisse von Patientenzielgruppen eingehen zu können, sofern sie alle Bedingungen des Artikels 5 (5) der IVDR einhalten. Sie definieren und dokumentieren somit eine neue Zweckbestimmung. Dafür müssen sie auch einige der in der IVDR geregelten Herstellerpflichten übernehmen, indem sie z.B. die Konformität mit den allgemeinen Sicherheits- und Leistungsanforderungen aus Anhang I der IVDR sicherstellen (Artikel 5 (5), Satz 1) [24], [25]. Um hier die MPBetreibV besser mit der IVDR zu harmonisieren, sieht der Referentenentwurf des Bundesgesundheitsministerium zur Änderung der MPBetreibV eine Aufhebung des § 4 Absatz 1 vor, sodass der Einsatz von Medizinprodukten entsprechend der Zweckbestimmung auch gemäß MPBetreibV dann nicht mehr geboten sein wird [10].
Davon unabhängig dürfen alle IVDs nur dann angewandt werden, wenn die Mängelfreiheit und Funktionsfähigkeit vor Anwendung gewährleistet werden kann (§ 4), z.B. durch Überprüfung von Verfallsdaten und der Prüfung von Instandhaltungsmaßnahmen vor dem Einsatz und durch Funktionskontrollen. Instandhaltungsmaßnahmen, wie Inspektionen und Wartungen, und Funktionstestungen müssen regelmäßig und fortwährend und unter Berücksichtigung der Herstellerangaben durchgeführt werden.
Gesundheitseinrichtungen mit regelmäßig mehr als 20 Beschäftigten müssen einen Beauftragten für Medizinproduktesicherheit benennen, der über eine Funktions-E-Mail-Adresse auf der Internetseite als zentrale Stelle innerhalb der Gesundheitseinrichtung als Kontaktperson, z.B. für Behörden oder für Meldungen von Risiken und Vorkommnissen, kontaktiert werden kann (§ 6). Der Beauftrage für Medizinproduktesicherheit koordiniert dabei auch die internen Prozesse zur Erfüllung der Meldepflicht gemäß Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV, siehe weiter unten).
Die MPBetreibV verpflichtet Einrichtungen, die laboratoriumsmedizinische Untersuchungen durchführen, dazu, vor Aufnahme der Tätigkeiten ein Qualitätssicherungssystem zur „Aufrechterhaltung der erforderlichen Qualität, Sicherheit und Leistung bei der Anwendung von In-vitro-Diagnostika sowie zur Sicherstellung der Zuverlässigkeit der damit erzielten Ergebnisse einzurichten“ (§ 9 (1), Satz 1). Eine ordnungsgemäße Qualitätssicherung wird angenommen, wenn Teil A der Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (RiliBÄK) beachtet wird (§ 9 (1), Satz 2).
RiliBÄK – die Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen
Die Anwendung der „Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen“ (RiliBÄK) ist seit 2001 in der MPBetreibV festgeschrieben. Am 30. Mai 2023 ist im Deutschen Ärzteblatt die aktuelle Version der RiliBÄK erschienen [5]. Sie ist als Kammer-Richtlinie weder Gesetz noch Verordnung; Ärztinnen und Ärzte sind jedoch gemäß § 5 der (Muster-)Berufsordnung für die in Deutschland tätigen Ärztinnen und Ärzte (MBO-Ä 1997) verpflichtet, an den von der Bundesärztekammer eingeführten Qualitätssicherungsmaßnahmen teilzunehmen [4]. Auch ist die Beachtung der RiliBÄK wegen der Vermutungswirkung in § 9 der MPBetreibV angeraten: Ihre Anwendung erfüllt die Forderung der MPBetreibV an ein geeignetes Qualitätssicherungssystem beim Betreiben und bei der Anwendung von IVDs. Abweichendes Handeln bedarf einer besonderen Begründung [4].
Ziel der Richtlinie ist es, die Qualität laboratoriumsmedizinischer Untersuchungen zu sichern, kontinuierlich zu verbessern und Risiken für Patienten und Anwender so gering wie möglich zu halten (Steckbrief RiliBÄK siehe Tabelle 6 [Tab. 6]) [7]. Dieses Ziel soll durch die Festlegung grundsätzlicher Anforderungen an die Struktur- und Prozessqualität laboratoriumsmedizinischer Untersuchungen in der Heilkunde, an das dafür erforderliche Qualitätsmanagement und an die dauerhafte Qualitätssicherung erreicht werden. Die Qualitätssicherung soll dabei risikobasiert sein. Die untersuchungsspezifische Risikobewertung ist zu dokumentieren.
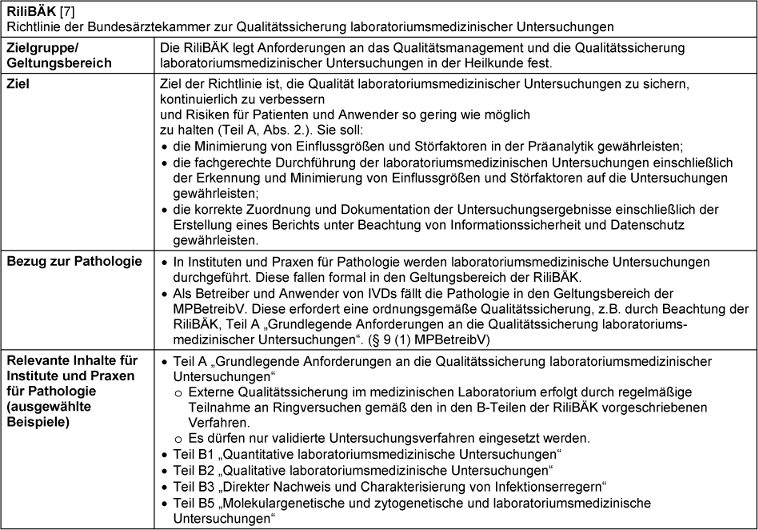
Die RiliBÄK beinhaltet die beiden für laboratoriumsmedizinische Untersuchungen relevanten Teile A und B. Teil A legt die grundlegenden Anforderungen an die Qualitätssicherung der Untersuchungen fest (z.B.: grundsätzliche Teilnahme an externen Ringversuchen (Teil A, Kapitel 8.2)). Teil A beschreibt Anforderungen für die Präanalytik (Einsendung), für die eigentliche Analytik, bis hin zur Postanalytik (Teil A, Kapitel 6). Es wird vorgeschrieben, dass nur validierte Untersuchungsverfahren eingesetzt werden dürfen. Die Validierung und die erhaltenen Ergebnisse müssen dokumentiert sein, ebenso wie das Untersuchungsverfahren, welches an den Arbeitsplätzen zur Verfügung stehen muss.
Teil B legt die untersuchungsspezifischen Anforderungen an die Ergebnisqualität und die dafür relevanten Grundsätze der Qualitätssicherung fest (z.B.: Anzahl der Ringversuche pro Kalenderjahr in Abhängigkeit der Untersuchung). Teil B gliedert sich in fünf „Spezielle Teile“ B1 bis B5:
- B1: Quantitative laboratoriumsmedizinische Untersuchungen
- B2: Qualitative laboratoriumsmedizinische Untersuchungen
- B3: Direkter Nachweis und Charakterisierung von Infektionserregern
- B4: Ejakulatuntersuchungen
- B5: Molekulargenetische und zytogenetische laboratoriumsmedizinische Untersuchungen
Für die Pathologie wären in erster Linie die speziellen Teile B1, B2, B3 und B5 zutreffend. Der Teil B5 wurde durch die Aktualisierung der RiliBÄK vom Mai 2023 aufgrund des technischen Fortschritts vollständig neu verfasst. Die Bundesärztekammer stellt auf ihrer Homepage ein Hilfedokument zur Verfügung, welches die „Antworten zu häufig gestellten Fragen zur RiliBÄK“ geben soll [6]. Nach einer aktuellen Entscheidung der Bundesärztekammer soll das Konzept einer möglichen Erweiterung der RiliBÄK um das Gebiet Pathologie erarbeitet werden, so dass alle Morphologie-basierten Verfahren (z.B. Immunhistochemie) Gegenstand der Richtlinie wären [11].
MPAMIV – die Medizinprodukte-Anwendermelde- und Informationsverordnung
Im Zuge der Einführung der IVDR und MDR und der Ablösung des Medizinproduktegesetzes (MPG) durch das Medizinprodukte-Durchführungsgesetz (MPDG) wurde auch die Medizinprodukte-Sicherheitsplanverordnung (MPSV) zum 26. Mai 2021 von der neuen nationalen Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV) abgelöst [33] (Abbildung 2 [Abb. 2]).
Die MPAMIV regelt das Meldeverfahren und den Informationsaustausch bei mutmaßlichen schwerwiegenden Vorkommnissen im Zusammenhang mit IVDs (Begriffsbestimmung siehe Tabelle 1 [Tab. 1]). Die MPAMIV gliedert sich in zwei Abschnitte (Steckbrief MPAMIV siehe Tabelle 7 [Tab. 7]). Abschnitt 1 „Anwendungsbereich; Meldeverfahren“ befasst sich mit der Meldung von mutmaßlichen schwerwiegenden Vorkommnissen und ist für Institute und Praxen für Pathologie als Betreiber und Anwender von IVD von Relevanz. Der Abschnitt 2 der MPAMIV legt den Informationsaustausch zwischen den zuständigen Behörden untereinander fest.

Mit der Regelung des Meldeverfahrens in Abschnitt 1 und der Meldepflicht für Betreiber und Anwender setzt die MPAMIV den in der IVDR (Artikel 82 Absatz 10) festgelegten Auftrag an die Mitgliedstaaten um. Mutmaßlich schwerwiegende Vorkommnisse im Zusammenhang mit Produkten, die auf dem Unionsmarkt bereitgestellt werden (CE-IVDs), sind vom Betreiber oder Anwender von IVD der zuständigen Bundesoberbehörde zu melden (§ 3). Dies gilt auch für Ärzte, die in Ausübung ihrer beruflichen Tätigkeit von solchen Vorkommnissen Kenntnis erlangen. Auch Patienten können eine Meldung direkt durchführen, sind hier jedoch im Gegensatz zu den Betreibern und Anwendern nicht dazu verpflichtet. Die Meldung erfolgt über das Deutsche Medizinprodukteinformations- und Datenbanksystem (DMIDS) nach § 86 des Medizinprodukterecht-Durchführungsgesetzes (MPDG) an die Bundesoberbehörde, entweder an das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM, [8]) oder an das Paul-Ehrlich-Institut (PEI, [27]). Die Koordination der dafür notwendigen Prozesse kann dabei sinnvollerweise zentral über den Beauftragten für Medizinproduktesicherheit (laut MPBetreibV, § 6) durchgeführt werden. Allerdings ist nicht vorgeschrieben, wer die Meldung durchführt. Das Regierungspräsidium Baden-Württemberg schlägt in einem Merkblatt zur Meldepflicht vor, dass jede Einrichtung für den Meldeprozess eine Verfahrensanweisung vorhält, die die Verantwortlichkeiten, die Meldefrist, die Internetadressen der Meldeformulare und die Regelung zur Aufbewahrung des betroffenen Produktes enthält [28].
Eine Meldung mutmaßlich schwerwiegender Vorkommnisse ist nur für Medizinprodukte vorgeschrieben, die auf dem Unionsmarkt bereitgestellt werden (CE-IVD) (IVDR, Artikel 82 (1)). Produkte, die von Gesundheitseinrichtungen nach IVDR, Artikel 5 (5), eigenhergestellt werden und ausschließlich innerhalb der Gesundheitseinrichtung eingesetzt werden (IH-IVD), sind von dieser Meldepflicht daher wohl nicht betroffen, da hier nach IVDR, Artikel 5 (5), keine Bereitstellung auf dem Unionsmarkt stattfinden darf (Begriffsbestimmung siehe Tabelle 1 [Tab. 1]).
Überwachungsmaßnahmen zum Medizinprodukterecht
Institute und Praxen für Pathologie agieren als Betreiber, Anwender und Eigenhersteller von In-vitro-Diagnostika und müssen je nach Funktion die entsprechenden gesetzlichen Anforderungen erfüllen (Abbildung 3a [Abb. 3]). Das europäische und nationale Medizinprodukterecht geben hierfür einen Rechtsrahmen vor, innerhalb dessen sich Institute und Praxen für Pathologie und deren Beschäftigte, in Abhängigkeit ihrer obigen Funktion, bewegen (Abbildung 3b + c [Abb. 3]). Nach Artikel 5 (5) der IVDR erhalten die zuständigen Behörden der Mitgliedstaaten Zugang zu den Gesundheitseinrichtungen, um deren Tätigkeiten zu überprüfen. Die Details dieses Betretungsrechts werden in § 79 Abs. 1 Nr. 1 und 2 des MPDG geregelt. Nach § 77 des MPDG sind die Länder für die Überwachung der Einhaltung der medizinproduktrechtlichen Vorschriften zuständig. Für ein bundeseinheitliches Vorgehen bei der qualitätsgesicherten und risikobasierten Überwachung durch die zuständigen Behörden der Länder wurde am 12. Juni 2023 auf Grundlage des § 89 MPDG die „Allgemeine Verwaltungsvorschrift zur Durchführung des Medizinprodukterechts (Medizinprodukterecht-Durchführungsvorschrift – MPRVwV)“ erlassen (Abbildung 3d [Abb. 3]) [9].
MPRVwV – die Medizinprodukterecht-Durchführungsvorschrift
Die im Juni 2023 erlassene „Allgemeine Verwaltungsvorschrift zur Durchführung des Medizinprodukterechts (Medizinprodukterecht-Durchführungsvorschrift, MPRVwV)“ richtet sich an die landesspezifischen Behörden, die für die Durchführung des Medizinprodukterechts zuständig sind, um ein bundeseinheitliches Vorgehen sicherzustellen [9]. Die Verwaltungsvorschrift legt fest, dass die Überwachungsmaßnahmen und Überwachungsintervalle auf Grundlage von Jahresplänen risikobasiert und qualitätsgesichert durchzuführen sind (Steckbrief MPRVwV siehe Tabelle 8 [Tab. 8]). Hierfür sollen die zuständigen obersten Landesbehörden Grundsätze und Anforderungen gemäß „§ 3 Grundsätze der Überwachung“ und „§ 5 System zur Qualitätssicherung“ festlegen. Für die Durchführung der Maßnahmen zur Qualitätssicherung muss in der Behörde mindestens eine qualifizierte Person abgestellt werden. Inspektionen sind in der Regel vor Ort durchzuführen und erfolgen durch Überprüfungen der produktbezogenen Unterlagen und Informationen, durch physische Kontrollen oder durch Laboruntersuchungen. Sie können routinemäßig gemäß Jahresplan, aber auch anlassbezogen durchgeführt werden. Sie dürfen sowohl angekündigt als auch unangekündigt durchgeführt werden. Dabei sollen sie die „Besonderheiten der Tätigkeiten in Betrieben und Einrichtungen berücksichtigen“ (§ 8).

Weitere Richtlinien und Normen
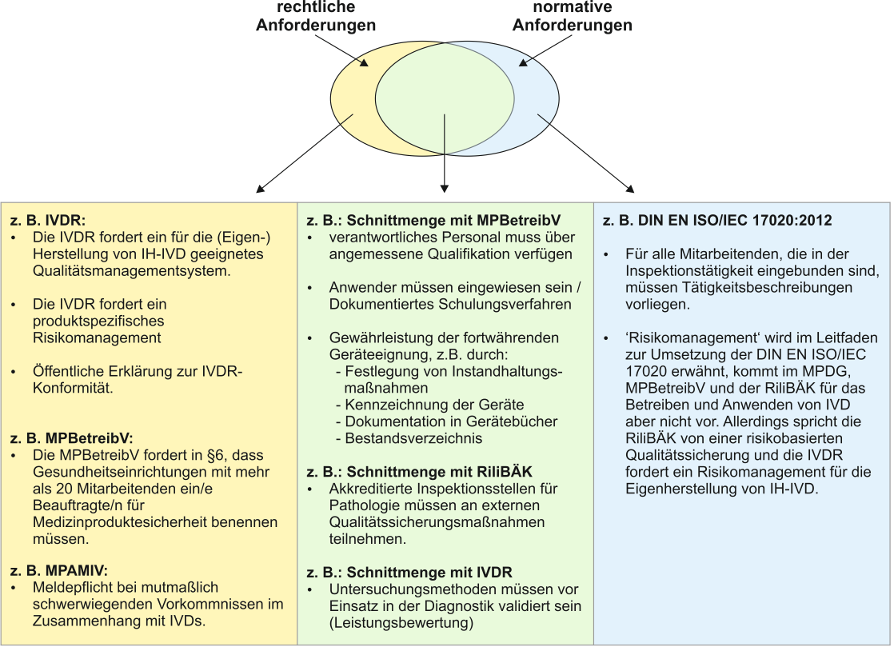
Institute und Praxen für Pathologie sind als Leistungserbringer im Gesundheitswesen gesetzlich dazu verpflichtet, ein Qualitätssicherungssystem (QMS) einzuführen und weiterzuentwickeln (Sozialgesetzbuch (SGB V) Neunter Abschnitt „Sicherung der Qualität der Leistungserbringung“ [29]). Die Qualitätsmanagement-Richtlinie (QM-RL) des Gemeinsamen Bundesausschusses (G-BA) beschreibt die Anforderungen für die Einführung und für die Umsetzung eines Qualitätsmanagements für medizinische Einrichtungen, die für gesetzlich versicherte Personen tätig sind [19]. Die QM-RL beinhaltet in Teil A die Rahmenbestimmungen, die gemeinsam für alle Sektoren gelten (wie z.B. Risiko-, Beschwerde- und Fehlermanagement), sowie die sektorspezifische Konkretisierungen, z.B. für die vertragsärztliche Versorgung (Teil B).
Die MPBetreibV fordert vor Aufnahme der Tätigkeiten „ein Qualitätssicherungssystem nach dem Stand der medizinischen Wissenschaft und Technik zur Aufrechterhaltung der erforderlichen Qualität, Sicherheit und Leistung bei der Anwendung von IVD sowie zur Sicherstellung der Zuverlässigkeit der damit erzielten Ergebnisse einzurichten“ (§ 9). Und auch die IVDR fordert ein geeignetes QMS, sofern die Einrichtung als Eigenhersteller von IH-IVDs agiert (Artikel 5 (5), b) [17]). Die IVDR fordert in Artikel 5 (5), c) weiter: „das Labor der Gesundheitseinrichtung entspricht der Norm EN ISO 15189 oder gegebenenfalls nationalen Vorschriften einschließlich nationaler Akkreditierungsvorschriften“ [17].
Normen spiegeln den Stand der Technik wider und definieren standardisierte und vereinheitlichte Anforderungen z.B. für Tätigkeiten, Produkte oder Managementsysteme. Sie sind jedoch nicht rechtsverbindlich, sofern deren Einhaltung nicht konkret gesetzlich vorgeschrieben ist. Die hier explizit genannte Norm EN ISO 15189 beschreibt die Anforderungen an die Qualität und Kompetenz medizinischer Laboratorien, jedoch beinhaltet diese Norm keine Anforderungen an die (Eigen-)Herstellung von Produkten, sodass sie folglich wohl nicht vollumfänglich dem Artikel 5 (5), b) genügt. Sie wurde 2022 strukturell und inhaltlich überarbeitet und liegt seit 2023 als deutsche Fassung DIN EN ISO 15189:2023-03 vor.
Die Deutsche Akkreditierungsstelle GmbH (DAkkS) hat den gesetzlichen Auftrag, als nationale Akkreditierungsbehörde der Bundesrepublik Deutschland auf Antrag die freiwillige Akkreditierung von Konformitätsbewertungsstellen durchzuführen (Verordnung (EG) Nr. 765/2008 [16], Akkreditierungsstellengesetzes (AkkStelleG) [20]). Mit einer Akkreditierung beurkundet die DAkkS, dass die auditierte Organisation ihre Arbeit „nach den Anforderungen international gültiger Normen, gesetzlicher Grundlagen und relevanter Regeln kompetent erbringen“ kann [15]. Eine Akkreditierung soll dem öffentlichen Interesse dienen, ist jedoch für die Gesetzeskonformität für Gesundheitseinrichtungen nicht verpflichtend. Durch eine Akkreditierung allein kann noch keine Gesetzeskonformität gewährleistet werden (Abbildung 4 [Abb. 4]). Die Akkreditierung medizinischer Labore erfolgt auf Grundlage der Norm DIN EN ISO 15189. Ergänzend dazu wird die RiliBÄK berücksichtigt [12].
In Deutschland sind über 100 Institute für Pathologie oder Neuropathologie durch die DAkkS nach DIN EN ISO/IEC 17020 als Inspektionsstellen akkreditiert (s. Suchergebnis in [14] mit folgendem Filter: Inspektionsstelle, Pathologie, aktiv; Treffer: 110), [23]. Im Mittelpunkt der Akkreditierung nach DIN EN ISO/IEC 17020 steht die sachverständige Beurteilung der Pathologin bzw. des Pathologen und damit die Bestätigung der fachlichen Kompetenz der Inspektionsstelle. Die Anforderungen der Norm DIN EN ISO 15189 müssen zur erfolgreichen Akkreditierung ebenfalls berücksichtigt werden [12]. Der Anforderungskatalog des Sektorkomitees Pathologie/Neuropathologie (Kennung: 71 SD 4 001 [13]) gab Interpretationen und Empfehlungen zur Norm DIN EN ISO/IEC 17020 und nahm in seiner Forderung zu Kapitel 7.1.4 der Norm DIN EN ISO/IEC 17020 Bezug zu den gesetzlichen Grundlagen: „Eine Übersicht über interne und externe Anweisungen, Verordnungen und Gesetze, die zu befolgen sind, muss existieren und deren Aktualisierung sichergestellt sein.“ Obwohl die Sektorkomitees und deren Hilfestellungen derzeit aufgrund von Umstrukturierungen innerhalb der DAkkS in den Expertenrat und Checklisten überführt werden, ist es sinnvoll, stets eine aktuelle Übersicht über die Gesetzeslage inklusive dem Medizinprodukterecht zu haben, auch wenn die Norm selbst dies nicht explizit fordert.
Fazit/Diskussion
Bei der ärztlich verantworteten Durchführung von diagnostischen Untersuchungsmethoden in der Pathologie kommen Medizinprodukte in Form von In-vitro-Diagnostika als essentieller Bestandteil zur Diagnosefindung zum Einsatz. In allen Funktionen – als Betreiber, Anwender und Eigenhersteller von IVD – steht für die Pathologie dabei die Patienten- und Anwendersicherheit und eine valide Diagnosefindung stets im Mittelpunkt. Um das hohe Niveau an Sicherheit und Gesundheitsschutz sicherzustellen, gibt es auf nationaler und europäischer Ebene gesetzliche und normative Grundlagen, die hierfür einen Gesetzesrahmen und den Stand der Technik bilden, indem sich Institute und Praxen für Pathologien bewegen.
Die Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. (AWMF) veröffentlicht bei (geplanten) Änderungen im Medizinprodukterecht regelmäßig Stellungnahmen. Sie stellt z.B. die Anforderungen an Gesundheitseinrichtungen dar, die mit Inkrafttreten der IVDR für die Eigenherstellung von IH-IVDs von Gesundheitseinrichtungen zu erfüllen sind, kommentiert diese und zeigt definitorische Lücken der Verordnung auf [1]. Die AWMF kommentierte auch den aktuellen Referentenentwurf des Bundesgesundheitsministeriums zur geplanten Änderung der Medizinproduktebetreiberverordnung (MPBetreibV) [3]. Dieser Entwurf sieht eine Erweiterung des Anwendungsbereichs vor, der in der zunehmenden Digitalisierung begründet ist. Nun soll in ihr auch der Umgang mit Medizinprodukte-Software geregelt werden. Dies ist im Einklang mit der Neuaufnahme von „Software“ in die Definitionen von „Medizinprodukt“ und „In-vitro-Diagnostikum“ der MDR und IVDR (Tabelle 1 [Tab. 1]). In den vorhergehenden Direktiven MDD und IVDD war Software noch kein Bestandteil der Definitionen. Darüber hinaus ist geplant § 4 Absatz 1 der MPBetreibV zu streichen. Dieser Absatz erlaubt aktuell den Einsatz von Medizinprodukten und IVDs nur entsprechend ihrer vom Hersteller gegebenen Zweckbestimmung, während die IVDR auch den Einsatz außerhalb der Zweckbestimmung erlaubt und somit Gesundheitseinrichtungen ermöglicht, auf die spezifischen Bedürfnisse von Patientenzielgruppen eingehen zu können (Artikel 5 (5)). Damit unterstreicht die geplante Aufhebung dieses Absatzes der MPBetreibV die Methodenfreiheit von Pathologinnen und Pathologen und stellt eine Harmonisierung mit der IVDR dar.
In-vitro-Diagnostika sind Produkte, die innerhalb einer ärztlich verantworteten Prozesskette in der Analytik eingesetzt werden, um eine Diagnosestellung zu ermöglichen. Es ist wichtig, hierbei das IVD als Medizinprodukt klar von der Durchführung des diagnostischen Verfahrens als Prozess und ärztliche Leistung in ärztlicher Verantwortung abzugrenzen [34], [35]. Dies geschieht aktuell nur ansatzweise im Hilfestellungsdokument MDCG-2023-1 der Medical Device Coordination Group (MDCG) für Gesundheitseinrichtungen [26]. Die MDCG und ihre Aufgaben sind in den Artikeln 98 und 99 der IVDR beschrieben. Sie hat die Aufgabe, Leitlinien für eine wirksame und harmonisierte Umsetzung der IVDR zu entwickeln, die jedoch rechtlich nicht bindend sind. Das Dokument MDCG-2023-1 soll für Gesundheitseinrichtungen Empfehlungen und Hilfestellungen beim Umgang mit In-house-in-vitro-Diagnostika (IH-IVD) geben, indem es die Anforderungen aus Artikel 5 (5) zusammenstellt und kommentiert. Hier wird beispielhaft der PCR-Mastermix als IVD genannt, nicht jedoch die PCR als Untersuchungsverfahren. Die Ad-hoc-Kommission In-vitro-Diagnostik der AWMF weist darauf hin, dass die Durchführung von diagnostischen Verfahren als Prozess und in ärztlicher Verantwortung kein IVD darstellt, sondern eine ärztliche Leistung, die nicht Regelungsgegenstand der IVDR ist, sondern deren Qualitätssicherung in Deutschland der ärztlichen Selbstverwaltung unterliegt, und fordert daher eine präzise Abgrenzung [2].
Fazit für die Praxis
- Als Betreiber und Anwender von CE-IVDs und als Hersteller, Betreiber und Anwender von IH-IVDs unterliegen Institute und Praxen für Pathologie dem europäischen und nationalen Medizinprodukterecht (siehe Abbildung 3a + b [Abb. 3]).
- Durch die Einführung der beiden EU-Verordnungen IVDR und MDR ergab sich im deutschen Medizinprodukterecht ein Bedarf an regulatorischer Anpassung (siehe Abbildung 2 [Abb. 2]).
- Institute und Praxen für Pathologie können auf Gesetzeskonformität überprüft werden. Die Kontrollen finden durch die länderspezifischen zuständigen Behörden statt (siehe Abbildung 3d [Abb. 3]).
- Normkonformität und DAkkS-Akkreditierung bilden eine gute Grundlage für die Einhaltung der gesetzlichen Anforderungen, aber sie beinhaltet nicht alle gesetzlichen Anforderungen an die Eigenherstellung (siehe Abbildung 4 [Abb. 4]) von IH-IVDs und an das Betreiben und Anwendung von CE- und IH-IVDs.
Anmerkungen
Interessenkonflikte
- Andy Kahles: Es besteht kein Interessenkonflikt.
- Hannah Goldschmid: Es besteht kein Interessenkonflikt.
- Anna-Lena Volckmar: Persönliche Honorare von AstraZeneca außerhalb der eingereichten Arbeit
- Daniel Kazdal: Es besteht kein Interessenkonflikt.
- Ulrich M. Gassner: Es besteht kein Interessenkonflikt.
- Peter Schirmacher: Es besteht kein Interessenkonflikt.
- Albrecht Stenzinger: Advisory Board/Vortag: AGCT, Aignostics, AstraZeneca, Bayer, BMS, Eli Lilly, Illumina, Incyte, Janssen, MSD, Novartis, Pfizer, Roche, Seattle Genetics, Takeda, Thermo Fisher; Grants: Bayer, BMS, Chugai, Incyte
- Karl-Friedrich Bürrig: Es besteht kein Interessenkonflikt.
- Vanessa Kääb-Sanyal: Es besteht kein Interessenkonflikt.
- Christa Flechtenmacher: Es besteht kein Interessenkonflikt.
- Michael Vogeser: Es besteht kein Interessenkonflikt.
- Monika Brüggemann: Es besteht kein Interessenkonflikt.
Literatur
[1] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. Stellungnahme der AWMF zur Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR). Berlin: AWMF; 2020. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/stellungnahmen/2020/ivdr-stellungnahme-august-2020.pdf[2] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. Stellungnahme der AWMF zu ärztlichen Leistungen in Labordiagnostik und Pathologie im Hinblick auf die IVDR. Berlin: AWMF; 2024. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/stellungnahmen/2023/20231215_AWMF_STN_Regulation_aerztlicher_Leistung_in_Labordiagnostik_u_Pathologie.pdf
[3] Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften e.V. Stellungnahme der AWMF zur Dritten Verordnung zur Änderung medizinprodukterechtlicher Vorschriften - Referentenentwurf zur Medizinproduktebetreiberverordnung (MPBtreibV) vom 19.10.2023. Berlin: AWMF; 2023. Available from: https://www.awmf.org/fileadmin/user_upload/dateien/stellungnahmen/2023/20231201_STN_Dritte_VO_zur_Aenderung_medizinprodukterechtlicher_Vorschriften.pdf
[4] Bundesärztekammer. (Muster-)Berufsordnung für die in Deutschland tätigen Ärztinnen und Ärzte - MBO-Ä 1997 - in der Fassung der Beschlüsse des 114. Deutschen Ärztetages 2011 in Kiel. Dtsch Arztebl. 2011;108(38):A-1980/B-1684/C-1668.
[5] Bundesärztekammer. Aktualisierung der Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen – Rili-BÄK. Dtsch Arztebl. 2023 May 30;120(21-22):A-994/B-858. DOI: 10.3238/arztebl.2023.rili_baek_QS_Labor
[6] Bundesärztekammer. Antworten zu häufig gestellten Fragen zur "Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (Rili-BÄK)". Stand: April 2019. 2019. Available from: https://www.bundesaerztekammer.de/fileadmin/user_upload/_old-files/downloads/pdf-Ordner/QS/FAQ-Rili-BAEK.pdf
[7] Bundesärztekammer. Richtlinie der Bundesärztekammer zur Qualitätssicherung laboratoriumsmedizinischer Untersuchungen (RiliBÄK). Gemäß des Beschlusses des Vorstands der Bundesärztekammer in seiner Sitzung am 18.10.2019, zuletzt geändert durch Beschlussfassung des Vorstands der Bundesärztekammer am 14.04.2023. Dtsch Arztebl. 2023;120(21-22). DOI: 10.3238/arztebl.2023.rili_baek_QS_Labor
[8] Bundesministerium für Arzneimittel und Medizinprodukte (BfArM). Formblatt für die Meldung von Vorkommnissen durch Betreiber und Anwender sowie Patienten oder deren Angehörige nach den §§ 3 und 4 der Medizinprodukte-Anwendermelde- und Informationsverordnung (MPAMIV). [last accessed 2024 Jan 19]. Bonn; BfArM. Available from: https://www2.bfarm.de/medprod/mpsv/
[9] Bundesministerium für Gesundheit (BMG). Allgemeine Verwaltungsvorschrift zur Durchführung des Medizinprodukterechts (Medizinprodukterecht-Durchführungsvorschrift – MPRVwV). Berlin/Bonn: 2023 Jun 12 [last accessed 2024 Jan 01]. Available from: https://www.verwaltungsvorschriften-im-internet.de/bsvwvbund_12062023_BMG.htm
[10] Bundesministerium für Gesundheit (BMG). Referentenentwurf - Dritte Verordnung zur Änderung medizinprodukterechtlicher Vorschriften. 2023. Available from: https://www.bundesgesundheitsministerium.de/service/gesetze-und-verordnungen/detail/verordnungen-zur-aenderung-medizinprodukterechtlicher-vorschriften.html
[11] Bundesverband deutscher Pathologen e.V.; Deutsche Gesellschaft für Pathologie e.V. Pressemitteilung. Erfolgreiche Grundsteinlegung: Erweiterung der Rili-BÄK um die Pathologie hat begonnen. Berlin: 2024 May 16. Available from: https://www.pathologie.de/_Resources/Persistent/f/d/6/2/fd623ecca1f11e8393be47df8b37c29545d22bd9/Pressemitteilung%20BDP%20DGP%20Rili-B%C3%84K%20Beiratssitzung%2015.04.2024%20%281%29.pdf
[12] Deutsche Akkreditierungsstelle (DAkkS). Medizinische Laboratoriumsdiagnostik 1 Pathologie – Fachbereich 3.5 im Überblick. [last accessed 2024 Jan 19]. Available from: https://www.dakks.de/de/fb-3.5.html
[13] Deutsche Akkreditierungsstelle (DAkkS). Sektorkomitee Pathologie/Neuropathologie. Anforderungen der DIN EN ISO/IEC 17020:2012 und technische Kriterien für deren Anwendung zur Akkreditierung in der Pathologie/Neuropathologie (71 SD 4 001, Revision: 1.6). Berlin: DAkksS; 2017. Available from: https://www.dakks.de/de/dokument-detail.html?id=anforderungen-der-din-en-iso-iec-17020-2012-und-technische-kriterien-fuer-deren-anwendung-zur-akkreditierung-in-der-pathologie-neuropathologie71-sd-4-001
[14] Deutsche Akkreditierungsstelle (DAkkS). Suchergebnis Akkreditierte Stellen. [last accessed: 2024 Jan 19]. Available from: https://www.dakks.de/de/akkreditierte-stellen-suchergebnis.html
[15] Deutsche Akkreditierungsstelle (DAkkS). Wir sind die DAkkS. Available from: https://www.dakks.de/de/home.html
[16] Europäisches Parlament. Verordnung (EG) Nr. 765/2008 des Europäischen Parlaments und des Rates vom 9. Juli 2008 über die Vorschriften für die Akkreditierung und Marktüberwachung im Zusammenhang mit der Vermarktung von Produkten und zur Aufhebung der Verordnung (EWG) Nr. 339/93 des Rates (Text von Bedeutung für den EWR). Amtsblatt der Europäischen Union. 2008 Aug 13;(L 218):30-47.
[17] Europäisches Parlament. Verordnung (EU) 2017/746 des Europäischen Parlaments und des Rates vom 5. April 2017 über In-vitro-Diagnostika und zur Aufhebung der Richtlinie 98/79/EG und des Beschlusses 2010/227/EU der Kommission (Text von Bedeutung für den EWR). Amtsblatt der Europäischen Union. 2017 May 05;(L 117):176-332.
[18] Eurpäisches Parlament. Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates vom 5. April 2017 über Medizinprodukte, zur Änderung der Richtlinie 2001/83/EG, der Verordnung (EG) Nr. 178/2002 und der Verordnung (EG) Nr. 1223/2009 und zur Aufhebung der Richtlinien 90/385/EWG und 93/42/EWG des Rates (Text von Bedeutung für den EWR). Amtsblatt der Europäischen Union. 2017 May 05;(L 117):1-175.
[19] Gemeinsamer Bundesausschuss. Richtlinie des Gemeinsamen Bundesausschusses über grundsätzliche Anforderungen an ein einrichtungsinternes Qualitätsmanagement für Vertragsärztinnen und Vertragsärzte, Vertragspsychotherapeutinnen und Vertragspsychotherapeuten, medizinische Versorgungszentren, Vertragszahnärztinnen und Vertragszahnärzte sowie zugelassene Krankenhäuser (Qualitätsmanagement-Richtlinie/QM-RL). Zuletzt geändert am 20. April 2023, in Kraft getreten am 21. Juli 2023. BAnz. 2023 Jul 07:B1. Available from: https://www.g-ba.de/downloads/62-492-3200/QM-RL_2023-04-20_iK-2023-07-21.pdf
[20] Gesetz über die Akkreditierungsstelle (Akkreditierungsstellengesetz - AkkStelleG). [last accessed 2024 Jan 19]. Available from: https://www.gesetze-im-internet.de/akkstelleg/
[21] Gesetz zur Anpassung des Medizinprodukterechts an die Verordnung (EU) 2017/745 und die Verordnung (EU) 2017/746 (Medizinprodukte-EU-Anpassungsgesetz - MPEUAnpG) 19. Wahlperiode. [last accessed 2024 Jan 19]. Available from: https://dip.bundestag.de/vorgang/.../255346
[22] Gesetz zur Durchführung unionsrechtlicher Vorschriften betreffend Medizinprodukte (Medizinprodukterecht-Durchführungsgesetz - MPDG). [last accessed 2024 Jan 19]. Available from: https://www.gesetze-im-internet.de/mpdg/
[23] Holl-Ulrich K, Hagel C, Köhler G, Flechtenmacher C. Akkreditierung in der Pathologie und Neuropathologie: Wege und Pitfalls [Accreditation in pathology and neuropathology: Paths and pitfalls]. Pathologie (Heidelb). 2022 Sep;43(5):338-45. DOI: 10.1007/s00292-022-01098-w
[24] Kahles A, Goldschmid H, Volckmar AL, Ploeger C, Kazdal D, Penzel R, Budczies J, Flechtenmacher C, Gassner UM, Brüggemann M, Vogeser M, Schirmacher P, Stenzinger A. Die Verordnung (EU) 2017/746 (IVDR) in der Praxis: Umsetzung von Anhang I in der Pathologie [Regulation (EU) 2017/746 (IVDR): practical implementation of annex I in pathology]. Pathologie (Heidelb). 2023 Nov;44(6):381-91. DOI: 10.1007/s00292-023-01231-3
[25] Kahles A, Goldschmid H, Volckmar AL, Plöger C, Kazdal D, Penzel R, Budczies J, Kempny G, Kazmierczak M, Flechtenmacher C, Baretton G, Weichert W, Horst D, Klauschen F, Gassner UM, Brüggemann M, Vogeser M, Schirmacher P, Stenzinger A. Struktur und Inhalt der EU-IVDR: Bestandsaufnahme und Implikationen für die Pathologie [Structure and content of the EU-IVDR: Current status and implications for pathology]. Pathologie (Heidelb). 2022 Sep;43(5):351-64. DOI: 10.1007/s00292-022-01077-1
[26] Medical Device Coordination Group. MDCG 2023-1: Guidance on the health institution exemption under Article 5(5) of Regulation (EU) 2017/745 and Regulation (EU) 2017/746. 2023. Available from: https://health.ec.europa.eu/latest-updates/mdcg-2023-1-guidance-health-institution-exception-under-article-55-regulation-eu-2017745-and-2023-01-10_en
[27] Paul-Ehrlich-Institut (PEI). Meldeformulare. [last accessed 2024 Jan 19]. Available from: https://www.pei.de/DE/arzneimittelsicherheit/ivd-vigilanz/meldeformulare/meldeformulare-node.html
[28] Regierungspräsidien Baden-Württemberg. Merkblatt: Meldepflicht der Betreiber und Anwender bei mutmaßlichen schwerwiegenden Vorkommnissen mit Medizinprodukten. Version 4, Stand: 02/2022. 2022. Available from: https://rp.baden-wuerttemberg.de/fileadmin/RP-Internet/Themenportal/Gesundheit/_DocumentLibraries/Documents/MerkblattMeldepflichtVorkommnissen.pdf
[29] Sozialgesetzbuch (SGB) Fünftes Buch (V) - Gesetzliche Krankenversicherung - (Artikel 1 des Gesetzes v. 20. Dezember 1988, BGBl. I S. 2477), Stand: Zuletzt geändert durch Art. 10 G v. 17.7.2023 I Nr. 191. [last accessed 2024 Jan 19]. Available from: https://www.gesetze-im-internet.de/sgb_5/
[30] Spitzenberger F, Patel J, Gebuhr I, Kruttwig K, Safi A, Meisel C. Laboratory-Developed Tests: Design of a Regulatory Strategy in Compliance with the International State-of-the-Art and the Regulation (EU) 2017/746 (EU IVDR [In Vitro Diagnostic Medical Device Regulation]). Ther Innov Regul Sci. 2022 Jan;56(1):47-64. DOI: 10.1007/s43441-021-00323-7
[31] Stenzinger A, Weichert W. Einfluss der neuen In-vitro-Diagnostik-Regulation (IVDR) der Europäischen Union auf die Pathologie. Was ist wichtig? [Impact of the novel in vitro diagnostic regulation (IVDR) of the European Union on pathology laboratories. What's important?]. Pathologie. 2020 Dec;41(Suppl 2):129-33. DOI: 10.1007/s00292-020-00867-9
[32] Verordnung über das Errichten, Betreiben und Anwenden von Medizinprodukten (Medizinprodukte-Betreiberverordnung - MPBetreibV). [last accessed 2024 Jan 19]. Available from: https://www.gesetze-im-internet.de/mpbetreibv/
[33] Verordnung über die Meldung von mutmaßlichen schwerwiegenden Vorkommnissen bei Medizinprodukten sowie zum Informationsaustausch der zuständigen Behörden (Medizinprodukte-Anwendermelde- und Informationsverordnung - MPAMIV). [last accessed 2024 Jan 19]. Available from: https://www.gesetze-im-internet.de/mpamiv/
[34] Vogeser M, Bruggemann M. Complex analytical procedures in diagnostic laboratories and the IVDR. Clin Chem Lab Med. 2020 Dec 11;59(3):457-8. DOI: 10.1515/cclm-2020-1775
[35] Vogeser M, Bruggemann M, Lennerz J, Stenzinger A, Gassner UM. Laboratory-Developed Tests in the New European Union 2017/746 Regulation: Opportunities and Risks. Clin Chem. 2022 Jan;68(1):40-2. DOI: 10.1093/clinchem/hvab215

