[Establishment of a globally accessible registry for auditory synaptopathy DFNB9 (OTOF)]
Barbara Vona 1,2Bernd Wollnik 2
Tobias Moser 1
Nicola Strenzke 1,3
1 University Medical Center Göttingen, Institute for Auditory Neuroscience, Göttingen, Germany
2 University Medical Center Göttingen Institute of Human Genetics, Göttingen, Germany
3 University Medical Center Göttingen, Department of Otolaryngology, Göttingen, Germany
Abstract
The autosomal recessive inherited auditory synaptopathy DFNB9 is caused by mutations in the OTOF gene and manifests itself in most cases as congenital deafness with absent ABR but preserved otoacoustic emissions. In rarer cases, residual hearing is present, albeit with significant impairment of speech comprehension, sometimes with pathological hearing fatigue and/or dependence of hearing on body temperature. The cause is a disturbance in the release and/or replenishment of the glutamate-filled vesicles at the synapse of the inner hair cell in the absence of the hair cell-specific protein otoferlin. Several animal studies and initial studies in humans show the feasibility in principle of restoring hearing through virus-mediated gene therapy.
In order to advance clinical research, we have established a “RedCap” database in which patients, their relatives and doctors can enter information on phenotype, genotype and inheritance patterns in pseudonymized format. The study scientists can also contact the participants – provided they have given their consent in electronic format.
The study is listed in ClinicalTrials.org and will enable a better overview of the incidence and clinical patterns of DFNB9, including genotype–phenotype correlations, the success of different treatments, as well as patient involvement in research.
Auditory synaptopathy DFNB9
The first described and most prevalent form of inherited auditory synaptopathy DFNB9 arises from mutations in the OTOF gene which encodes the otoferlin protein [1]. Since its initial description, nearly 300 disease-causing variants in OTOF have been identified. The prevalence of DFNB9 varies between population and reaches up to 5% of cases of congenital deafness [2]. DFNB9 is an auditory synaptopathy, characterized by the presence of otoacoustic emissions despite absent ABRs and stapedial reflexes. In this constellation, deafness may remain undetected by newborn hearing screening when OAE are used as a diagnostic tool. The majority of DFNB9 patients are profoundly deaf. In the few patients with significant residual hearing, speech comprehension is limited by auditory fatigue, and benefit from hearing aids is limited. In contrast, the causal functional deficit can be successfully bypassed when using a cochlear implant for hearing rehabilitation (reviewed in [3]).
Otoferlin, a multi-C2-domain protein, plays a crucial role in the synaptic transmission between sensory inner hair cells and spiral ganglion neurons. Its functions include calcium sensing for fusion [4], [5], [6], vesicle replenishment [7], [8], [9] and exocytosis-endocytosis coupling [10], [11], [12]. Due to the clearly defined defect and the relatively mild degenerative changes [13], OTOF has emerged as a promising target for gene therapy development (review in [14]). Soon after successful pre-clinical studies demonstrating the feasibility of virus-mediated gene therapy in Otof knockout mouse models [15], [16], [17], five clinical studies (NCT05901480, ChiCTR2200063181, NCT05821959, NCT05788536, SENS-501) were initiated. The published preliminary results are promising [18], [19], but data regarding the long-term efficacy and safety are still missing (reviewed in [20], [21]).
The OTOF registry
Inspired by our continued basic science research on Otoferlin and the translational potential, we have now developed an otoferlin registry to promote translational studies. We need a broader knowledge on genotype-phenotype correlations, on the natural history of the disease, and on the success of different treatment modalities. We need to be able to re-contact to ask for follow-up data and to recruit them to studies, e.g. to specifically characterize temperature sensitive hearing loss associated with DFNB9 and auditory fatigue. We would like to be able to directly contact patients, and to inform them about new discoveries, to involve them in the planning of new studies, and to establish connections with and among them.
The registry operates on a locally installed REDCap database hosted by the working group for Biometry, Data Management and Informatics in Clinical Studies at the University Medical Center Göttingen. To ensure data security, the structure comprises two databases, separating patient-identifiable information (blue in Figure 1 [Fig. 1]) from clinical and genetics data (green in Figure 1 [Fig. 1]). The consent procedure begins by accessing the Otoferlin Registry section through the Institute for Auditory Neuroscience website (https://www.auditory-neuroscience.uni-goettingen.de/otoferlin_registry_en.html). This page contains study information and consent forms for adults, parents, adolescents, and children. Following completion of the fully digital consent procedure and provision of personal information, participants receive a participant-specific link and access code via an encrypted email for personal access to the registry questionnaire. This structure guarantees the strict separation of personal and medical information. The structured questionnaire comprises questions regarding the genetic test results, disease course, family history, and hearing rehabilitation. Genetic and audiometric tests should be uploaded as a variety of possible file types such as PDF or JPG after removal of personal identifiers. All data is checked for completeness and adherence to data protection rules by the study coordinator. Participants are able to review and amend their submitted data and submit updates or additional audiometry over time using the same personal link. They also receive copies of all transmitted consent forms and registry data for their records. A second pseudonymization is applied prior to publication of data.
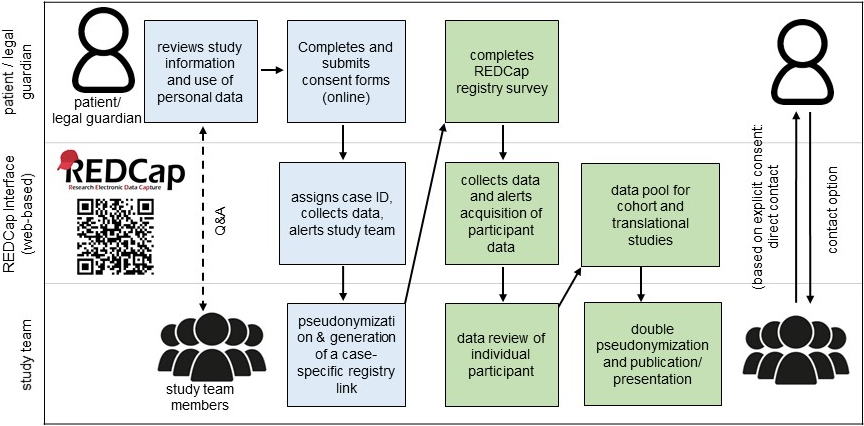
To our knowledge, this gene-specific registry represents the first for an isolated form of hearing impairment. The bilingual implementation attracts German- and English-speaking participants.
We currently plan an update to track the results of gene therapy. This will enable an independent evaluation that supplements, compares and follows up upon the standardised evaluation in the study protocols and after the corresponding treatments have been approved. Additionally, we aim to foster community engagement by reaching out to participants who have indicated their willingness to be recontacted. We envision the registry serving as a platform for advancing patient-centred research, advocacy and collaborative efforts. All participants who registered so far have explicitly signed up for the option to be recontacted and to receive educational or informational materials. One exciting prospect on our agenda is the organization of an otoferlin symposium for participants and their families. This event will provide an opportunity to discuss otoferlin research collaboratively, leveraging the expertise of patients and their families, who we believe are the true experts of otoferlin-associated synaptopathy and the primary stakeholders in the development of future approved otoferlin therapies.
Finally, we underscore the importance of genetic testing in the routine diagnostic work-up of patients with hearing impairment for which a hereditary cause may be possible. A genetic diagnosis, established through the identification of variants in OTOF, serves as the sole eligibility criterion for participation in the registry, but is also crucial for the participation in clinical trials, especially regarding causative treatment approaches.
In conclusion, we foresee the registry as a powerful tool for enhancing our understanding of the variants and clinical characteristics associated with OTOF-associated hearing impairment. It will facilitate genotype-phenotype correlations and enable real-time assessment of therapeutic modalities. Moreover, the registry will serve as a platform for engaging the OTOF patient community, fostering collaboration for patient advocacy and facilitating our own OTOF clinical trials. The study design can also serve as a template for similar projects.
Further information and resources
To learn more about the otoferlin registry or for patient enrolment, please visit https://www.auditory-neuroscience.uni-goettingen.de/otoferlin_registry_en.html. This study was approved by the Ethics Committee of the University Medical Center Göttingen (Approval: 17/8/22) and is registered on ClinicalTrials.gov under ID NCT05946057.
Prof. Dr. Nicola Strenzke (nicola.strenzke@med.uni-goettingen) is available for clinical questions on the diagnosis and treatment of DFNB9, and Prof. Dr. Bernd Wollnik (bernd.wollnik@med.uni-goettingen.de) for general questions on genetic diagnostics.
Notes
Conference presentation
This contribution was presented at the 26th Annual Conference of the German Society of Audiology and published as an abstract [22].
Acknowledgements
We thank members of the Institute of Human Genetics Leipzig for productive discussions in the early planning stages of our registry. We thank Thomas Asendorf and members of the clinical trials unit and ethics commission at the University Medical Center Göttingen for the constructive collaboration.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy EXC 2067/1-390729940 (to T.M. and B.W.), the DFG Heisenberg program STR 1027/5-1 grant 406266759 (to N.S.) and the DFG VO 2138/7-1 grant 469177153 (to B.V.).
Competing interests
The authors declare that they have no competing interests.
References
[1] Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999 Apr;21(4):363-9. DOI: 10.1038/7693[2] Vona B, Rad A, Reisinger E. The Many Faces of DFNB9: Relating OTOF Variants to Hearing Impairment. Genes (Basel). 2020 Nov 26;11(12):1411. DOI: 10.3390/genes11121411
[3] Moser T, Starr A. Auditory neuropathy--neural and synaptic mechanisms. Nat Rev Neurol. 2016 Mar;12(3):135-49. DOI: 10.1038/nrneurol.2016.10
[4] Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006 Oct 20;127(2):277-89. DOI: 10.1016/j.cell.2006.08.040
[5] Johnson CP, Chapman ER. Otoferlin is a calcium sensor that directly regulates SNARE-mediated membrane fusion. J Cell Biol. 2010 Oct 4;191(1):187-97. DOI: 10.1083/jcb.201002089
[6] Michalski N, Goutman JD, Auclair SM, Boutet de Monvel J, Tertrais M, Emptoz A, Parrin A, Nouaille S, Guillon M, Sachse M, Ciric D, Bahloul A, Hardelin JP, Sutton RB, Avan P, Krishnakumar SS, Rothman JE, Dulon D, Safieddine S, Petit C. Otoferlin acts as a Ca2+ sensor for vesicle fusion and vesicle pool replenishment at auditory hair cell ribbon synapses. Elife. 2017 Nov 7;6:e31013. DOI: 10.7554/eLife.31013
[7] Pangrsic T, Lasarow L, Reuter K, Takago H, Schwander M, Riedel D, Frank T, Tarantino LM, Bailey JS, Strenzke N, Brose N, Müller U, Reisinger E, Moser T. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010 Jul;13(7):869-76. DOI: 10.1038/nn.2578
[8] Vogl C, Cooper BH, Neef J, Wojcik SM, Reim K, Reisinger E, Brose N, Rhee JS, Moser T, Wichmann C. Unconventional molecular regulation of synaptic vesicle replenishment in cochlear inner hair cells. J Cell Sci. 2015 Feb 15;128(4):638-44. DOI: 10.1242/jcs.162099
[9] Strenzke N, Chakrabarti R, Al-Moyed H, Müller A, Hoch G, Pangrsic T, Yamanbaeva G, Lenz C, Pan KT, Auge E, Geiss-Friedlander R, Urlaub H, Brose N, Wichmann C, Reisinger E. Hair cell synaptic dysfunction, auditory fatigue and thermal sensitivity in otoferlin Ile515Thr mutants. EMBO J. 2016 Dec 1;35(23):2519-35. DOI: 10.15252/embj.201694564
[10] Duncker SV, Franz C, Kuhn S, Schulte U, Campanelli D, Brandt N, Hirt B, Fakler B, Blin N, Ruth P, Engel J, Marcotti W, Zimmermann U, Knipper M. Otoferlin couples to clathrin-mediated endocytosis in mature cochlear inner hair cells. J Neurosci. 2013 May 29;33(22):9508-19. DOI: 10.1523/JNEUROSCI.5689-12.2013
[11] Jung S, Maritzen T, Wichmann C, Jing Z, Neef A, Revelo NH, Al-Moyed H, Meese S, Wojcik SM, Panou I, Bulut H, Schu P, Ficner R, Reisinger E, Rizzoli SO, Neef J, Strenzke N, Haucke V, Moser T. Disruption of adaptor protein 2μ (AP-2μ) in cochlear hair cells impairs vesicle reloading of synaptic release sites and hearing. EMBO J. 2015 Nov 3;34(21):2686-702. DOI: 10.15252/embj.201591885
[12] Kroll J, Jaime Tobón LM, Vogl C, Neef J, Kondratiuk I, König M, Strenzke N, Wichmann C, Milosevic I, Moser T. Endophilin-A regulates presynaptic Ca2+ influx and synaptic vesicle recycling in auditory hair cells. EMBO J. 2019 Mar 1;38(5):e100116. DOI: 10.15252/embj.2018100116
[13] Stalmann U, Franke AJ, Al-Moyed H, Strenzke N, Reisinger E. Otoferlin Is Required for Proper Synapse Maturation and for Maintenance of Inner and Outer Hair Cells in Mouse Models for DFNB9. Front Cell Neurosci. 2021 Jul 14;15:677543. DOI: 10.3389/fncel.2021.677543
[14] Kleinlogel S, Vogl C, Jeschke M, Neef J, Moser T. Emerging Approaches for Restoration of Hearing and Vision. Physiol Rev. 2020 Oct 1;100(4):1467-525. DOI: 10.1152/physrev.00035.2019
[15] Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, Boutet de Monvel J, Hardelin JP, Hauswirth WW, Avan P, Petit C, Safieddine S, Lustig LR. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci U S A. 2019 Mar 5;116(10):4496-501. DOI: 10.1073/pnas.1817537116
[16] Al-Moyed H, Cepeda AP, Jung S, Moser T, Kügler S, Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. 2019 Jan;11(1):e9396. DOI: 10.15252/emmm.201809396
[17] Rankovic V, Vogl C, Dörje NM, Bahader I, Duque-Afonso CJ, Thirumalai A, Weber T, Kusch K, Strenzke N, Moser T. Overloaded Adeno-Associated Virus as a Novel Gene Therapeutic Tool for Otoferlin-Related Deafness. Front Mol Neurosci. 2021 Jan 7;13:600051. DOI: 10.3389/fnmol.2020.600051
[18] Lv J, Wang H, Cheng X, Chen Y, Wang D, Zhang L, Cao Q, Tang H, Hu S, Gao K, Xun M, Wang J, Wang Z, Zhu B, Cui C, Gao Z, Guo L, Yu S, Jiang L, Yin Y, Zhang J, Chen B, Wang W, Chai R, Chen ZY, Li H, Shu Y. AAV1-hOTOF gene therapy for autosomal recessive deafness 9: a single-arm trial. Lancet. 2024 May 25;403(10441):2317-25. DOI: 10.1016/S0140-6736(23)02874-X
[19] Qi J, Tan F, Zhang L, Lu L, Zhang S, Zhai Y, Lu Y, Qian X, Dong W, Zhou Y, Zhang Z, Yang X, Jiang L, Yu C, Liu J, Chen T, Wu L, Tan C, Sun S, Song H, Shu Y, Xu L, Gao X, Li H, Chai R. AAV-Mediated Gene Therapy Restores Hearing in Patients with DFNB9 Deafness. Adv Sci (Weinh). 2024 Mar;11(11):e2306788. DOI: 10.1002/advs.202306788
[20] Moser T, Chen H, Kusch K, Behr R, Vona B. Gene therapy for deafness: are we there now? EMBO Mol Med. 2024 Apr;16(4):675-7. DOI: 10.1038/s44321-024-00058-6
[21] Strenzke N. A cure for deafness? Med. 2024 Apr 12;5(4):285-7. DOI: 10.1016/j.medj.2024.02.007
[22] Strenzke N, Vona B, Wollnik B, Moser T. Etablierung eines weltweit zugänglichen Registers für die auditorische Synaptopathie DFNB9 (OTOF). In: Deutsche Gesellschaft für Audiologie e.V. 26. Jahrestagung der Deutschen Gesellschaft für Audiologie. Aalen, 06.-08.03.2024. Düsseldorf: German Medical Science GMS Publishing House; 2024. Doc150. DOI: 10.3205/24dga150

